
SÍNDROME DEL X FRAGIL (ó Síndrome de Martin-Bell.)
")
Más de 12.000 personas padecen en la Argentina el Síndrome del cromosoma X frágil, pero sólo hay 250 casos diagnosticados. Muchas más son las personas portadoras de la mutación causante. Es el responsable del 17% de los casos de retraso intelectual y suele confundirse con autismo, pero un diagnóstico correcto permitiría tomar importantes decisiones para mejorar la calidad de vida de los afectados y de sus familias.

DEBAJO DE LA NÚMERO 10
En el festejo de uno de sus golazos contra el Real Madrid, en mayo de 2009, Lionel Messi dejó ver, debajo de la camiseta del Barcelona, una remera blanca con la leyenda “Síndrome X frágil”. Esta acción solidaria del actual Número Uno del fútbol mundial permitió que millones de personas, que seguían a través de sus televisores ese súper clásico en todo el mundo, registrasen ese nombre hasta entonces prácticamente desconocido.

El compromiso desinteresado del crack con las familias afectadas por el síndrome X frágil surgió a través del insistente pedido de una madre residente en Barcelona, que removió cielo y tierra para que llegar a él y hacerlo partícipe de la tarea de hacer conocida esta problemática, que así como afectó a su hijo, probablemente está afectando a muchas otras familias sin que estas siquiera lo sepan.
Gracias a ese gesto de Messi y de la visibilidad del trabajo de la Asociación, las consultas a los sitios relacionados con X frágil se multiplicaron en todo el mundo, y seguramente muchos pudieron tomar conciencia de la enfermedad que estaba afectando a alguno de los suyos y, sobre todo, de que no están solos frente a ella.
¿De qué se trata?
El Síndrome del X frágil es la primera causa de retraso mental heredable o bien la mayor causa de discapacidad intelectual hereditaria. Es un desorden dominante monogénico X ligado, o sea que hay un gen, que se ubica en el Cromosoma X, que es el que al estar alterado, lo genera observándose principalmente en varones, los cuales tienen un fenotipo (aspecto físico) característico.
El grado de afectación es variable. Puede oscilar de un retraso mental leve a un retraso grave asociado a autismo. Incluso es posible que, en ocasiones, sólo se exprese como un trastorno del aprendizaje o problemas![]() emocionales, en cuyo caso, se habla de síndrome X Frágil de funcionamiento elevado.
emocionales, en cuyo caso, se habla de síndrome X Frágil de funcionamiento elevado.
Si bien el retraso mental es el síntoma más característico, las manifestaciones de este síndrome son complejas.
La sospecha clínica se basa en un fenotipo físico, cognitivo y conductual y el diagnóstico se confirma mediante el estudio genético molecular mediante una extracción de sangre.
El nombre se debe a que esta asociado con un SITIO FRAGIL O SITIO FRAXA ubicado en la banda 27 del brazo largo del cromosoma X (Xq27).
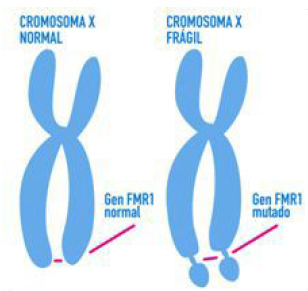
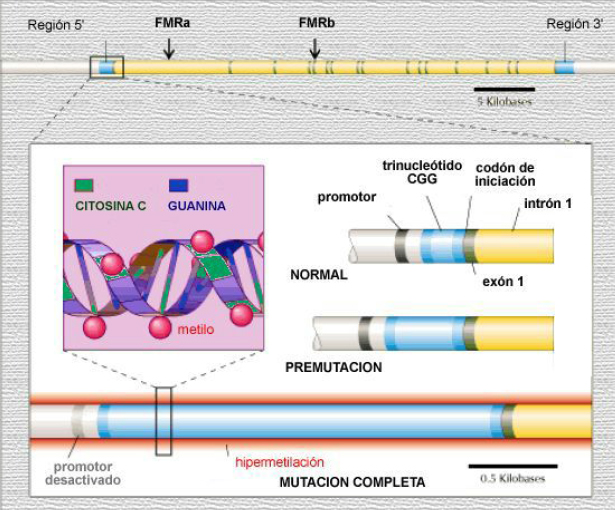
El sitio FRAGIL o sitio FRAXA es una anormalidad como una “constricción o sitio frágil” que se visualiza haciendo citogenética y allí se aloja![]() el gen afectado, denominado FMR-1 (Fragile X Mental Retardation 1), el cual codifica para una proteína denominada FMPR, pero en la región 5’ no traducida de dicho gen ( a izquierda de la secuencia de nucleótidos codificantes del gen) está la secuencia repetitiva de trinucleótidos (CGG)n con un n que va generalmente de 6 a 56 unidades en la población normal, en cambio para quienes padecen esta patología, existe una expansión considerable de este triplete repetitivo, con un n mayor a 200, lo que genera que el gen FMR-1 NO se exprese, no se traduzca entonces a su proteína (la FMRP) y eso origina el cuadro del síndrome, donde lo mas sobresaliente es el retraso mental.
el gen afectado, denominado FMR-1 (Fragile X Mental Retardation 1), el cual codifica para una proteína denominada FMPR, pero en la región 5’ no traducida de dicho gen ( a izquierda de la secuencia de nucleótidos codificantes del gen) está la secuencia repetitiva de trinucleótidos (CGG)n con un n que va generalmente de 6 a 56 unidades en la población normal, en cambio para quienes padecen esta patología, existe una expansión considerable de este triplete repetitivo, con un n mayor a 200, lo que genera que el gen FMR-1 NO se exprese, no se traduzca entonces a su proteína (la FMRP) y eso origina el cuadro del síndrome, donde lo mas sobresaliente es el retraso mental.
La función de la FMRP es unir el ARNm de cualquier gen transcripto y trasladarlo del núcleo al citoplasma.
La proteína FMRP en un caso normal, se expresa máximamente en el cerebro, en las gónadas (testículos y ovarios), en cartílagos e hígado. La carencia de dicha proteína en diferente grado, (según la mutación) acarreará los síntomas mayormente a nivel de los tejidos, órganos y glándulas citadas.
En el cerebro aparentemente juega un rol en el reestablecimiento de la sinapsis del árbol dendrítico
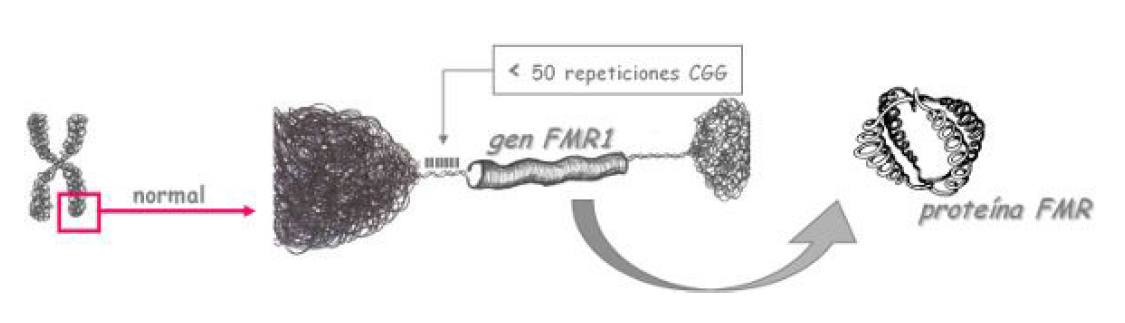
Hay quienes no tienen la mutación completa sino que son portadores de lo que se llama PREMUTACION, que es cuando la n varía entre 56 y 200. Estos individuos, cuando son hombres, se los llama HOMBRES T o TRANSMISORES. Son fenotípicamente y citogeneticamente normales, pero la secuencia repetitiva de trinucleotidos (CGG)n en ellos tiene la capacidad de alargarse al pasar de generación en generación. Aquí entra el concepto de MUTACION DINAMICA, es así porque se alarga y agrava la mutación al pasar de generación en generación, con lo cual el riesgo mayor lo tienen los descendientes al convertirse la PREMUTACION en una MUTACION COMPLETA. Con esto, describimos un desorden heredado pero NO en su forma clásica (no se hereda según las Leyes de Mendel).
Este tipo de mutacion también introduce el concepto de FENOMENO DE ANTICIPACION, el cual se basa en que la expansión del triplete (CGG) al pasar de una generación a otra, nos avisa que a medida que descendemos en la genealogía, va a aparecer mayor numero de casos graves, descendientes cada vez mas enfermos.

Cuando la n oscila entre 6 y 56 (lo normal para la cantidad de tripletes CGG), o entre 56 y 200, el grado de metilación de las CITOSINAS (C) en un cluster CG aledaño al tándem (CGG)n, es BAJO Y EL GEN FMR-1 SE EXPRESA, TRADUCIENDOSE LUEGO EN LA PROTEINA QUE CODIFICABA, LA FMRP
Cuando se tiene la mutacion completa porque la n supera las 200 copias del triplete (CGG), la isla CG mencionada, está HIPERMETILADA en su CITOSINA (C) con lo cual el gen FMR-1 NO PUEDE EXPRESARSE y NO HABRA PROTEINA FMRP, por ende no habrá quien cumpla sus funciones.
Prevalencia de la enfermedad:
Las mujeres al ser 46 XX genotípicamente tienen una chance del 50 % de estar afectadas clínicamente, si portan el X que tiene el gen mutado. El otro 50 % es tener el X que aloja![]() el gen normal, (porque quedó inactivo el X con gen mutado) y serán sanas. (**ver NOTA de Genética al pie; opcional)
el gen normal, (porque quedó inactivo el X con gen mutado) y serán sanas. (**ver NOTA de Genética al pie; opcional)
Los hombres, al ser 46 XY NO tienen esa chance, y son mas afectados, ya que si heredan el X con gen con mutación completa (y no con la premutación) NO tienen opción y siempre se expresará la mutación, lo que implica NO contar con la proteína FMRP y todas sus consecuencias.
En un principio, según el método citogenético, se estaba sobreestimando la prevalencia ya que contaba con baja sensibilidad, dando falsos positivos y decíamos que las prevalencias eran:
- Hombres afectados: 1:1250
- Mujeres afectadas: 1:2500 ( la mitad de chances que el hombre al ser a la mitad la frecuencia de afectadas)
Se debía a que ese método de visualización reconoce otros sitios frágiles que no tienen nada que ver con el de esta enfermedad.
Hoy día mediante estudios moleculares (mas específicos que las tinciones citogenéticas) se recalcularon los sitios FRAXA arrojando prevalencias menores para ambos sexos, pero siempre se mantiene la relación 2 a 1 entre hombres afectados vs mujeres afectadas. Por lo cual, las pruebas basadas en ADN, identifican carriers (o portadores) con premutación y con la mutacion completa.
La prevalencia para la mutación completa en la población general es de 1: 4500.
- Hombres afectados: 1:4500
- Mujeres afectadas: 1:9000 (la mitad de chances que el hombre al ser a la mitad la frecuencia de afectadas) asumiendo el 50 % de carriers o portadoras.
Premutación: Hombres: 1:1000 y Mujeres: 1: 400
En la Argentina, sólo 250 familias se encuentran diagnosticadas; el resto desconoce ser portadores de este síndrome hereditario, y por lo tanto seguirán teniendo hijos afectados.
Aproximadamente 1/3 de las portadoras de la mutación completa del gen X frágil pueden padecer algún grado de déficit intelectual que puede ir de leve a grave.
Según estudios realizados en países desarrollados, 1 de cada 250 mujeres y 1 de cada 700 varones son portadores del gen que lo produce, y el 80 a 90% de los individuos afectados por el síndrome de X Frágil permanecen sin diagnóstico.
En España se estima que la frecuencia es de 1 por cada 4.000 varones en la población general, una portadora por cada 800 y un portador por cada 5.000 nacidos vivos.
Patrón de herencia:
- Es desconocido el mecanismo que causa la expansión de la mutación.
- El número de los trinucleótidos (n) (CGG), repetidos aumenta de padres a hijos (Mutacion Dinámica), con lo cual el riesgo del X frágil aumenta de una generación a otra.
- Todas las mutaciones completas derivan de premutaciones.
- El riesgo de expansión de la premutación en una mujer carrier a la mutacion completa es alta y depende del número de repetidos CGG.
Excepciones: Puede haber pacientes con Síndrome del X Frágil y con deleciones o mutaciones nonsense (sin sentido)
¿Cómo se manifiesta clínicamente el SXF?
Características somáticas del recién nacido:
- Peso corporal ligeramente superior al promedio de controles normales
- Apariencia normal
Características somáticas en el hombre:
- Cara angosta y alargada (apariencia adulta acromegálica).Mentón prominente.
- Orejas prominentes, con pabellones auriculares grandes.
- Testículos grandes (macroorquidismo): mayor a 25 ml de volumen cada testículo en adultos y mas de 2 ml de vol en cada testículo en prepuberes. En más del 90 % de los casos se observa post pubertad. El excesivo desarrollo testicular se debe a que en las gónadas (testículos en este caso) la proteína FMRP se debería expresar mucho y no lo hace pro estar mutado su gen.
- Subtipos asociados con sobrecaimiento y fenotipo Prader Willi con obesidad extrema.
- Displasia del tejido conectivo general, por alteración a nivel del Colágeno (orejas, hiperextensibilidad de articulaciones, pie plano, prolapso de válvula mitral lo que se acusa en soplo cardíaco en el ECG y auscultación), piel suave como el terciopelo.
- Bajo tono muscular
- Estrabismo.
- Otitis de repetición.


Características de Comportamiento, Cognición y Neuropsicología en el hombre:
- Aparición tardía del lenguaje
- Hiperextensibilidad.
- Hiperactividad y déficit de atención.
- Trastorno de ansiedad.
- Trastorno emocional y de conducta.
- Extrema sensibilidad.
- A veces agresión.
- Síntomas similares al autismo.
- Defectos en la memoria auditiva de corto-termino.
- Defecto en capacidades visual-espacial.
- Defecto en la coordinación visual-motora.
- Defectos en habilidades aritméticas.
- Epilepsia.
- Convulsiones.
Niveles de expresión de la FMRP correlacionan fuertemente con el IQ de hombres mosaicos pre/full mutación y hombre con la mutación completa (hipermetilada la C) o parcialmente metilada la C.
Características clínicas en mujeres con la mutación completa:
- Menos afectadas en todos los niveles (inactivación del X al azar), el 50 % presenta retraso mental con un déficit intelectual que va desde incapacidad para aprender con un IQ normal hasta retraso mental severo.
- IQ promedio 80 – 90.
- Algunas células producen FMRP (la proteína del gen FMR-1)
- Rango amplio de severidad.
- Características faciales similares a las de los hombres afectados.
- Alteraciones del comportamiento mucho menos severas que en los hombres.
Características de los carriers (portadores) de la premutación: Hombres y Mujeres:
Clínicamente normales tanto los hombres T (transmisores) como las mujeres carriers o portadoras de la premutación.
¿Cómo se diagnostica el SXF?
Indicaciones para el diagnostico:
- Individuos de cualquier sexo con retardo mental de desarrollo o autismo, especialmente si tienen:
- Características similares al SXF.
- Historia familiar de SXF.
- Parientes con retardo mental sin diagnóstico.
- Individuos que buscan un consejo para la reproducción y tienen:
- Historia familiar de SXF.
- Historia familiar de retardo mental sin diagnóstico.
- Fetos de una madre que es carrier para el SXF.
- Individuos con una prueba citogenética de X frágil discordante con su fenotipo.
Diagnostico clínico y diferencial:
- Fenotipo e historia familiar de retraso mental X-ligado ya que el SXF representa el 20 % de los retrasos mentales X ligados pero es el más frecuente de los heredados.
- Descartar el autismo y otros síndromes asociados a retraso mental.
Diagnostico citogenético:
- Utilizar dos o más medios de inducción (inducir los sitios frágiles en el cromosoma X), ya que la expresión de los sitios frágiles es variable.
- En Hombres: analizar 50 - 100 células en metafase (usando Linfocitos de sangre periférica).
- En Mujeres: analizar 75 - 150 células en metafase (usando Linfocitos de sangre periférica).
Si da mayor o igual al 2 % el numero de celulas que tienen los sitios FRAXA, se considera DIAGNOSTICO PRESUNTIVO, porque pueden no ser los sitios fragiles específicos de este Síndrome, ya que aledaños a los FRAXA hay otros que pudieron haber sido inducidos con esta técnica.
- Incluir un análisis cromosómico estándar (Cariotipo Standard), para descartar otro caso.
Un Test (+) NO es específico de X frágil.
Diagnóstico Molecular:
- Análisis del ADN: prueba especifica para la expansión FMR-1
- Cuando se desconoce la etiología del retardo mental: la evaluación genética debe incluir análisis citogenético de rutina. (Anormalidades cromosomitas constitucionales vistas en un cariotipo )
- Individuos en riesgo por historia familiar de SFX: con la prueba de ADN es suficiente.
- Prenatal: después de detectar una madre carrier.
Pruebas de laboratorio que hacen al diagnóstico por biología molecular:
Se busca la premutación y si hay mutacion completa. Se cuantifica el numero de repeticiones del triplete (CGG)
- Southern Blott: con las Enzimas de Restricción Eco R1; Hindi; BC/I y PsfI.
- PCR.
Diagnostico prenatal:
Indicado después de detectar a la madre como carrier (+)
Citogenética:
- Vellosidades corionicas
- Amniocitos.
- Sangre fetal
EL RIESGO DE QUE UNA PREMUTACION SE CONVIERTA EN MUTACION COMPLETA:
En Hombres: es muy pequeña.
En Mujeres: depende del tamaño de la premutación:
** Menos de 70 copias 10 %
** Entre 70 – 90 copias 50 – 70 %
** Mayor que 90 copias 90 %
Estudios con Anticuerpos contra la proteína FMRP:
Western Blott: Tamaño de la proteína y abundancia (importante en casos de pacientes con SXF por deleciones o mutaciones nonsense (sin sentido).
Inmunohistoquímica: con un extendido de linfocitos de sangre periférica o células de fluido amniótico.
El número de células (+): debido a mosaicismo para repetidos no metilados pequeños.
Tratamiento del SXF:
El SXF NO tiene cura y todos los niños afectados requieren:
- Mejorar la concentración y disminuir la agresividad, en el caso de estar presente, son los objetivos principales del tratamiento en la niñez temprana.
- Terapia del habla, del lenguaje y ocupacional, por medio del Centro educativo del paciente
- Técnicas conductuales junto con terapias de coordinación motora fina y gruesa, pueden apaciguar un poco al paciente.
- El uso de medicación psicotrópica es una herramienta útil en muchos casos. Particularmente en niños de edad preescolar, las medicaciones estimulantes, como el metilfenidato, dextroanfetamina y adderall se asocian a menudo con un incremento de la irritabilidad y solo son eficaces en el 60 % de los casos.
- La clonidina, que tiene acción apaciguante, ayuda a controlar los síntomas de hiperactividad y agresividad en la mayoría de los niños con SXF.
Hay que realizar un cuidadoso seguimiento con electrocardiogramas (ECG) periódicos si se emplea algún tipo de medicación psicotrópica.

L. Laura Ocera
**NOTA para quienes deseen ampliar conceptos de genética o ya sepan:
El fenómeno epigenético llamado inactivación del cromosoma X ha intrigado a los científicos durante décadas.
En 1961 Mary Lyon formuló la hipótesis de que dicha inactivación del X se lleva a cabo al azar en fases precoces del periodo embrionario, y queda fijada una vez que se establece. Según esta hipótesis, todas las células hijas procedentes de una célula en la que se ha producido la inactivación tendrán el mismo patrón de inactivación que la célula original
En la inmensa mayoría de los genes se expresan los dos alelos. Sin embargo, esto no es posible en los cromosomas sexuales, ya que el número de cromosomas X e Y difiere entre los dos sexos. M. Barr y E. Bertram observaron por primera vez en el núcleo, un corpúsculo condensado distinto del nucleolo y se dieron cuenta de que las gatas normales poseen un único corpúsculo condensado mientras que los gatos no muestran ninguno. Estos investigadores se refirieron a este corpúsculo como cromatina sexual y desde entonces se ha denominado corpúsculo de Barr. En 1999, Mary Lyon sugirió que este corpúsculo de Barr representaba un cromosoma X inactivo el cual se enrolla en las hembras de manera compacta, tomando la estructura conocida como heterocromatina, una forma condensada y por tanto visible de la cromatina. Investigaciones recientes al microscopio revelan que los extremos del corpúsculo de Barr están en estrecha proximidad, formando un anillo.
Varias evidencias apoyan la hipótesis de Lyon. No obstante la desactivación NO es absoluta muchas veces, expresándose en torno a un 15% de los genes, explicándose así enfermedades de dosis génica como el Síndrome de Klinefellter (47 XXY) que trataremos en otro articulo.
Primero, los varones 47 XXY (Síndrome de Klinefelter) poseen un corpúsculo de Barr mientras que las mujeres 45 X0 (Síndrome de Turner) no presentan ninguno. Segundo, las personas con un número anormal de cromosomas X tienen un corpúsculo de Barr menos que cromosomas X poseen por célula: las mujeres 47 XXX presentan 2 corpúsculos de Barr y las 48 XXXX presentan tres. Demostración de la hipótesis de Lyon La prueba directa de la hipótesis de Lyon se produjo cuando, en mujeres normales (46 XX), los citólogos identificaron el corpúsculo de Barr como el cromosoma X. Las pruebas genéticas también apoyan la hipótesis de Lyon: las hembras heterocigóticas para un locus del cromosoma X muestran un patrón peculiar de expresión fenotípica. Se sabe ahora que en los seres humanos el cromosoma X es inactivado en cada célula alrededor del decimosegundo día de vida fetal y además el azar determina qué cromosoma X se inactiva en cada célula. A partir de este momento, el mismo cromosoma X permanece como un corpúsculo de Barr para futuras generaciones celulares. En lugar de ser hembras típicamente heterocigotas, son hemicigotas en cada célula para uno u otro de los alelos del cromosoma X.
La inactivacion es para reservar la dosis génica ya que desde los óvulos y espermatozoides, las mujeres son X y los hombres son Y ó X respectivamente y el embrión debe ser o 46 XX o 46 XY para tener una dosis cromosomita normal.
Importancia de la compensación de la dosis génica: El cromosoma X tiene unos 1000 genes que no están presentes en el pequeño cromosoma Y. Las hembras tienen el doble de copias de estos genes ligados al X y expresarían el doble de los transcritos de estos genes si no existiera un mecanismo para corregir este desequilibrio. Sin embargo, no tener un cromosoma Y no es un problema para las hembras ya que los pocos genes que hay en este cromosoma sólo son necesarios para el desarrollo de los machos. Este desajuste se corrige mediante un proceso llamado compensación de dosis, que hace que, a pesar de esta diferencia, las células femeninas y masculinas tengan cantidades equivalentes de las proteínas codificadas por genes del cromosoma X. La compensación de dosis supera diferencias de sexo en la relación esperada de la dosis de genes autosómicos con la dosis de genes del cromosoma X. Las relaciones de dosis son importantes ya que los productos de los genes ligados a X deben interactuar con los productos de los genes autosómicos en las diferentes vías metabólicas y del desarrollo, y las cantidades de producto para genes sensibles a dosis fundamentales están bajo regulación estrecha.

